Links
Is the 1000 genomes data available in genome browsers?
Answer:
1000 Genomes Project data is available at both Ensembl and the UCSC Genome Browser.
More information on accessing 1000 Genomes Project data in genome browsers can be found on the Browser page.
Ensembl provides consequence information for the variants. The variants that are loaded into the Ensembl database and have consequence types assigned are displayed on the Variation view. Ensembl can also offer consequence predictions using their Variant Effect Predictor (VEP).
You can see individual genotype information in the Ensembl browser by looking at the Individual Genotypes section of the page from the menu on the left hand side.
Related questions:
Are the 1000 genomes variants in dbSNP?
Answer:
The 1000 Genomes Project SNPs and short indels were all submitted to dbSNP and longer structural variants to the DGVa.
Where possible, release VCF files contain the appropriate IDs in the ID column, such as dbSNP rs IDs.
The archives contain variants discovered by the final phase of the 1000 Genomes Project (phase 3) and also by the preliminary pilot and phase 1 stages of the project. As methods were developed during the project, phase 3 represents the final data set.
Related questions:
Are all the genotype calls in the 1000 Genomes Project current release VCF files bi-allelic?
Answer:
No. While bi-allelic calling was used in earlier phases of the 1000 Genomes Project, multi-allelic SNPs, indels, and a diverse set of structural variants (SVs) were called in the final phase 3 call set. More information can be found in the main phase 3 publication from the 1000 Genomes Project and the structural variation publication. The supplementary information for both papers provides further detail.
In earlier phases of the 1000 Genomes Project, the programs used for genotyping were unable to genotype sites with more than two alleles. In most cases, the highest frequency alternative allele was chosen and genotyped. Depth of coverage, base quality and mapping quality were also used when making this decision. This was the approach used in phase 1 of the 1000 Genomes Project. As methods were developed during the 1000 Genomes Project, it is recommended to use the final phase 3 data in preference to earlier call sets.
Related questions:
Are there any assemblies available for the 1000 Genomes samples?
Answer:
The 1000 genomes project did not create any assemblies from the genome sequence data it generated.
The Gerstein Lab at Yale University created a diploid version of the NA12878 sequence, which is available from the Gerstein website under NA12878_diploid. When used, groups should cite AlleleSeq: analysis of allele-specific expression and binding in a network framework, Rozowsky et al., Molecular Systems Biology 7:522.
Related questions:
Are there any FASTA files containing 1000 Genomes variants or haplotypes?
Answer:
We do not provide FASTA files annotated for 1000 Genomes variants. You can create such a file with a VCFtools Perl script called vcf-consensus.
An example set of command lines would be:
#Extract the region and individual of interest from the VCF file you want to produce the consensus from
tabix -h ftp://ftp.1000genomes.ebi.ac.uk/vol1/ftp/release/20110521/ALL.chr17.phase1_release_v3.20101123.snps_indels_svs.genotypes.vcf.gz 17:1471000-1472000 | perl vcf-subset -c HG00098 | bgzip -c > HG00098.vcf.gz
#Index the new VCF file so it can be used by vcf-consensus
tabix -p vcf HG00098.vcf.gz
#Run vcf-consensus
cat ref.fa | vcf-consensus HG00098.vcf.gz > HG00098.fa
You can get more support for VCFtools on their help mailing list.
Related questions:
Are there any scripts or APIs for use with the 1000 Genomes data sets?
Answer:
There are a number of tools available in the Tools page of the 1000 Genomes Browser.
Our data is in standard formats like SAM and VCF, which have tools associated with them. To manipulate SAM/BAM files look at SAMtools for a C based toolkit and links to APIs in other languages. To interact with VCF files look at VCFtools which is a set of Perl and C++ code.
We also provide a public MySQL instance with copies of the databases behind the 1000 Genomes Ensembl browsers. These databases are described on our public instance page.
Related questions:
Are there any statistics about how much sequence data has been generated by the 1000 Genomes Project?
Answer:
Statistics about how much data the 1000 Genomes Project produced are accessible in several different ways. Information on some of the formats used for this information is available on the FTP site.
For raw data, a sequence.index file contains base and read counts for each of the active FASTQ files.
During the 1000 Genomes Project, summery statistics were provided in a sequence indices directory, which is now located with historical data from the project. This contains four summary files, two exome and two low coverage. Both of these analysis groups have a .stats file providing numbers of runs withdrawn for different reasons, base count and coverage statistics for each study, population level summaries and a stats.csv file which provides a comparison to the previous index in terms of number of runs, bases and similar metrics. Since late 2012, the 1000 Genomes Project also produced analysis.sequence.index files, which only consider Illumina runs of 70bp read length or longer, and also have statistics files.
For the aligned data all BAM and CRAM files have BAS files associated with them. These contain read group level statistics for the alignment. We also provide this in a collected form in alignment index files. The alignment indices for the alignments of the 1000 Genomes Project data to GRCh38 are available on the FTP site. There is also an historic alignment indices directory, which contains a .hsmetrics file with the results of the Picard tool CalculateHsMetrics for all the exome alignments and summary files, which compare statistics between old and new alignment releases during the 1000 Genomes Project.
Related questions:
Are there torrents available for the 1000 Genomes data sets?
Answer:
There are no official torrents of the 1000 Genomes Project data sets.
Related questions:
Can I access 1000 Genomes data with Globus Online?
Answer:
The 1000 Genomes FTP site is available as an end point in the Globus Online system. In order to access the data you need to sign up for an account with Globus via their signup page. You must also install the Globus Connect Personal software and setup a personal endpoint to download the data too.
The 1000 Genomes data is hosted at the EMBL-EBI end point called “Shared EMBL-EBI public endpoint”. Data from our FTP site can then be found under the 1000g directory within the EMBL-EBI public end point.
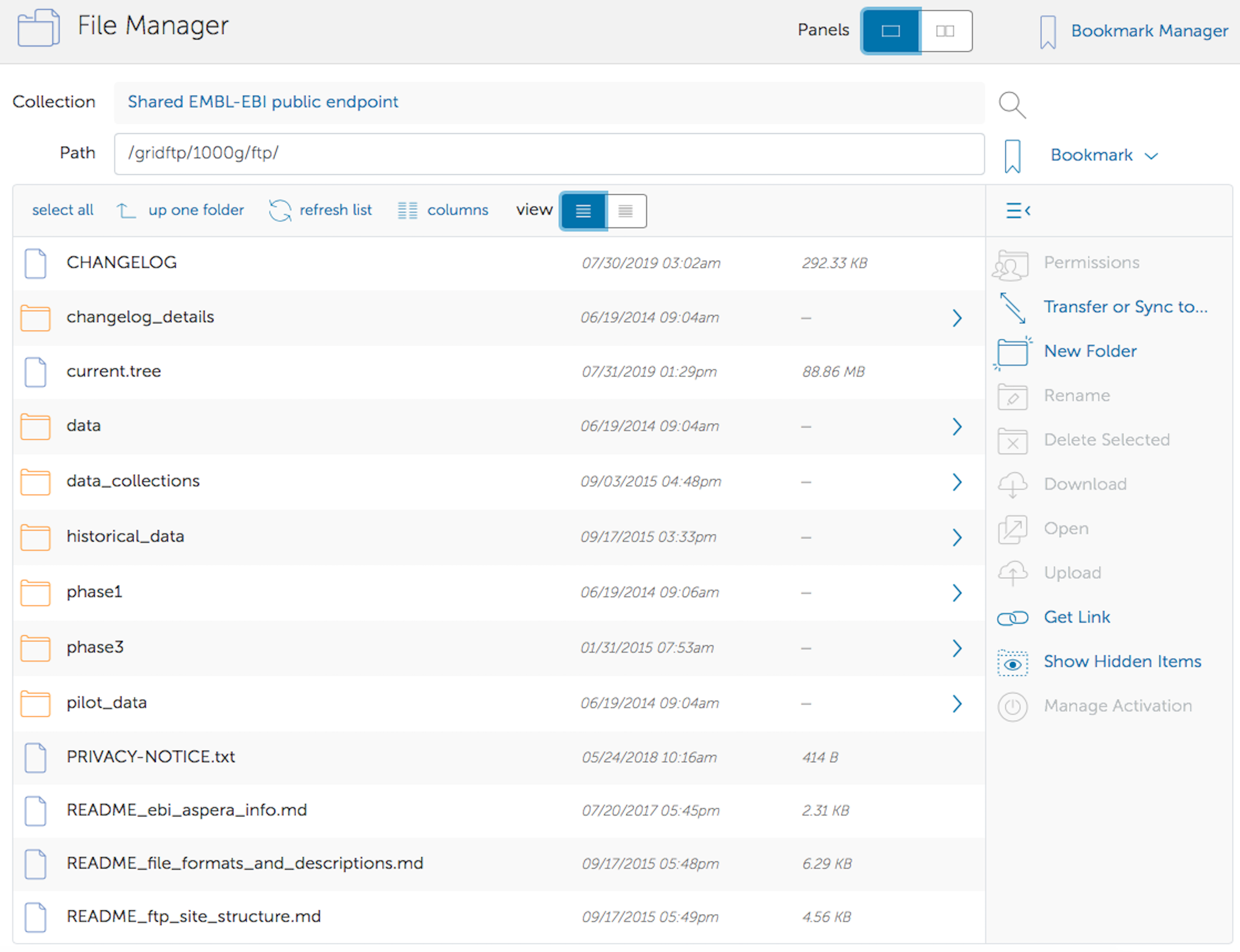
When you have setup your personal end point you should be able to start a transfer using their web interface.
The Globus website has support for setting up accounts, and installing the globus personal connect software.
Related questions:
Can I access the databases associated with the 1000 Genomes browser?
Answer:
We provide a public MySQL instance with copies of the databases behind the 1000 Genomes Project Ensembl browser. These databases are described on our public instance page. More information about the browsers and their history can be found on the browsers page.
Related questions:
Can I BLAST against the 1000 Genomes data sets?
Answer:
The 1000 Genomes raw sequence data represents more then 30,000x coverage of the human genome and there are no tools currently available to search against the complete data set. You can, however, use the Ensembl or NCBI BLAST services and then use these results to find 1000 Genomes Project variants in dbSNP.
Related questions:
Can I convert VCF files to PLINK/PED format?
Answer:
We provide a VCF to PED tool to convert from VCF to PLINK PED format. This tool has documentation for both the web interface and the Perl script.
An example Perl command to run the script would be:
perl vcf_to_ped_converter.pl -vcf ftp://ftp.1000genomes.ebi.ac.uk/vol1/ftp/release/20110521/ALL.chr13.phase1_integrated_calls.20101123.snps_indels_svs.genotypes.vcf.gz
-sample_panel_file ftp://ftp.1000genomes.ebi.ac.uk/vol1/ftp/release/20110521/phase1_integrated_calls.20101123.ALL.sample_panel
-region 13:32889611-32973805 -population GBR -population FIN
Related questions:
Can I find the genomic position for a list of dbSNP rs numbers?
Answer:
This can be done using Ensembl’s Biomart.
This YouTube video gives a tutorial on how to do it.
The basic steps are:
- Select the Ensembl Variation Database
- Select the Homo sapiens Short Variants (SNPs and indels excluding flagged variants) dataset
- Select the Filters menu from the left hand side
- Expand the General Variant Filters section
- Check the Filter by Variant Name (e.g. rs123, CM000001) [Max 500 advised] box
- Add your list of rs numbers to the box or browse for a file which contains this list
- Click on the Results Button in the headline section
- This should provide you with a table of results which you can also download in Excel or CSV format
If you would like the coordinates on GRCh38, you should use the main Ensembl site, however if you would like the coordinates on GRCh37, you should use the dedicated GRCh37 site.
Related questions:
Can I get 1000 Genomes data on the Amazon Cloud?
Answer:
At the end of the 1000 Genomes Project, a large volume of the 1000 Genomes data (the majority of the FTP site) was available on the Amazon AWS cloud as a public data set.
At the end of the 1000 Genomes Project, the IGSR was established and the FTP site has been further developed since the conclusion of the 1000 Genomes Project, adding additional data sets. The Amazon AWS cloud reflects the data as it was at the end of the 1000 Genomes Project and does not include any updates or new data.
You can find more information about how to use the data in the Amazon AWS cloud on our AWS help page.
Related questions:
Can I get cell lines for 1000 Genomes Samples?
Answer:
The 1000 Genomes Project sample cell lines are available for others to use. You can source them from the Coriell Cell Repository from the NHGRI collection.
A full listing of all the 1000 Genomes populations available from Coriell is available.
Related questions:
Can I get genotypes for a specific individual/population from VCF files?
Answer:
Either the Data Slicer or using a combination of tabix and VCFtools allows you to sub sample VCF files for a particular individual or list of individuals.
The Data Slicer, described in more detail in the documentation, has both filter by individual and population options. The individual filter takes the individual names in the VCF header and presents them as a list before giving you the final file. If you wish to filter by population, you also must provide a panel file which pairs individuals with populations, again you are presented with a list to select from before being given the final file, both lists can have multiple elements selected.
To use tabix you must also use a VCFtools Perl script called vcf-subset. The command line would look like:
tabix -h ftp://ftp-trace.ncbi.nih.gov/1000genomes/ftp/release/20100804/ALL.2of4intersection.20100804.genotypes.vcf.gz 17:1471000-1472000 | perl vcf-subset -c HG00098 | bgzip -c /tmp/HG00098.20100804.genotypes.vcf.gz
Related questions:
Can I get image files for any of the 1000 Genomes sequencing runs?
Answer:
The image files produced by next generation sequencing runs are always very big. The production centres do not submit them to the archives and they are not available to downstream users.
Related questions:
Can I get individual genotype information from browser.1000genomes.org?
Answer:
The 1000 genomes browser at browser.1000genomes.org but all data is accessible from the Ensembl browser at grch37.ensembl.org You can see individual genotype information in the browser by looking at the Sample Genotypes section of the a variant page. This can be reached from the menu on the left hand side of the page. You can find a particular variant by putting its rs number in the search box visible at the top right hand corner of every browser page.
Related questions:
Can I get phenotype, gender and family relationship information for the 1000 Genomes samples?
Answer:
For the 1000 Genomes Project, due to the freely available nature of the data, no phenotype information was collected for any of the samples. All donors were over 18 and declared themselves to be healthy at the time of collection. We do provide a sample spreadsheet and a pedigree file which contain ethnicity and gender for 1000 Genomes samples.
Related questions:
Can I search the website?
Answer:
You can search the website.
Every page on {{site.url}} has a search box in the top right hand corner. This allows you to find information anywhere on the IGSR website.
Related questions:
Can I volunteer to be part of the 1000 genomes project?
Answer:
The 1000 Genomes Project is not accepting volunteers to be sequenced. More information about how samples were recruited please see the About page.
Another large scale resequencing project that does still have rounds of recruitment is the Personal Genomes Project
Related questions:
Is the data for the pilot study still available?
Answer:
All the pilot data remains on our ftp site under the pilot_data directory EBI/NCBI. The variants which are discussed in the pilot paper can also be found on the ftp site EBI/NCBI.
Please note these data are all mapped to the NCBI36 human reference.
Related questions:
Do I need a password to access 1000 genomes data?
Answer:
All the 1000 genomes information is freely available without passwords.
There are 2 main sources for our raw and analysis data our ftp site which has 2 mirrored locations EBI|NCBI. These are accessible using both ftp and a udp protocol called ascp which is available freely from aspera. More information about these can be found on the data access page. Variant calls are also loaded into ensembl databases and can be browsed from here and the mysql database can be both accessed via our Public mysql instance and downloaded from our ftp site
Related questions:
Do I need permission to use the 1000 Genomes data in my own scientific research?
Answer:
The 1000 Genomes data is made available according to the Fort Lauderdale Agreement. The IGSR data use statement can be found on our data disclaimer page.
Related questions:
How can I get the allele frequency of my variant?
Answer:
Our VCF files contain global and super population alternative allele frequencies. You can see this in our most recent release. For multi allelic variants, each alternative allele frequency is presented in a comma separated list.
An example info column which contains this information looks like
1 15211 rs78601809 T G 100 PASS AC=3050;AF=0.609026;AN=5008;NS=2504;DP=32245;EAS_AF=0.504;AMR_AF=0.6772;AFR_AF=0.5371;EUR_AF=0.7316;SAS_AF=0.6401;AA=t|||;VT=SNP
If you want population specific allele frequencies you have three options: * For a single variant you can look at the population genetics page for a variant in our browser. This gives you piecharts and a table for a single site. * For a genomic region you can use our allele frequency calculator tool which gives a set of allele frequencies for selected populations * If you would like sub population allele frequences for a whole file, you are best to use the vcftools command line tool.
This is done using a combination of two vcftools commands called vcf-subset and fill-an-ac
An example command set using files from our phase 1 release would look like
grep CEU integrated_call_samples.20101123.ALL.panel | cut -f1 > CEU.samples.list
vcf-subset -c CEU.samples.list ALL.chr13.integrated_phase1_v3.20101123.snps_indels_svs.genotypes.vcf.gz | fill-an-ac |
bgzip -c > CEU.chr13.phase1.vcf.gz
</pre>
Once you have this file you can calculate your frequency by dividing AC (allele count) by AN (allele number).
Please note that some early VCF files from the main project used LD information and other variables to help estimate the allele frequency. This means in these files the AF does not always equal AC/AN. In the phase 1 and phase 3 releases, AC/AN should always match the allele frequency quoted.
Related questions:
How do I cite IGSR?
Answer:
When citing IGSR in general, please cite our Nucleic Acids Research database paper. For further details on citing specifc data sets, please see the appropriate data reuse policy. If you have any questions, please contact us at info@1000genomes.org.
Related questions:
How do I cite the 1000 genomes project?
Answer:
When citing the 1000 Genomes Project in general please use the final phase 3 paper, A global reference for human genetic variation, The 1000 Genomes Project Consortium, Nature 526, 68-74 (01 October 2015) doi:10.1038/nature15393. This paper is published under the creative commons Attribution-NonCommercial-ShareAlike 3.0 Unported licence, please feel free to share and redistribute the paper appropriately.
Related questions:
How do I contact you?
Answer:
If you want to ask us a question please email info@1000genomes.org
Related questions:
How do I find out about new 1000 genomes releases?
Answer:
We announce all new data releases on the front page of our website.
You can also follow these announcements on rss and twitter We have a public email list all announcements are posted to 1000announce@1000genomes.org
If you want to ask us a question please email info@1000genomes.org
Related questions:
How do I get a sub-section of a BAM file?
Answer:
There are two ways to get subsections of our BAM files.
The first is to use the Data Slicer tool from our browser which is documented here. This tool gives you a web interface requesting the URL of any BAM file and the genomic location you wish to get a sub-slice for. This tool also works for VCF files.
The second it to use samtools on the command line, e.g
samtools view -h ftp://ftp.1000genomes.ebi.ac.uk/vol1/ftp/phase1/data/HG00154/alignment/HG00154.mapped.ILLUMINA.bwa.GBR.low_coverage.20101123.bam 17:7512445-7513455
Samtools supports streaming files and piping commands together both using local and remote files. You can get more help with samtools from the samtools help mailing list
Related questions:
How do I get a sub-section of a VCF file?
Answer:
There are two ways to get a subset of a VCF file.
The first is to use the Data Slicer tool from our browser which is documented here. This tool gives you a web interface requesting the URL of any VCF file and the genomic location you wish to get a sub-slice for. This tool also works for BAM files. This tool also allows you to filter the file for particular individuals or populations if you also provide a panel file.
The second method is using tabix on the command line. e.g
tabix -h ftp://ftp.1000genomes.ebi.ac.uk/vol1/ftp/release/20100804/ALL.2of4intersection.20100804.genotypes.vcf.gz 2:39967768-39967768
Specifications for the VCF format, and a C++ and Perl tool set for VCF files can be found at vcftools on sourceforge
Please note that all our VCF files using straight intergers and X/Y for their chromosome names in the Ensembl style rather than using chr1 in the UCSC style. If you request a subsection of a vcf file using a chromosome name in the style chrN as shown below it will not work.
tabix -h ftp://ftp.1000genomes.ebi.ac.uk/vol1/ftp/release/20100804/ALL.2of4intersection.20100804.genotypes.vcf.gz chr2:39967768-39967768
Related questions:
- Can I convert VCF files to PLINK/PED format?
- What is the depth of coverage of your Phase1 variants?
- Can I find the genomic position for a list of dbSNP rs numbers?
- Can I get genotypes for a specific individual/population from VCF files?
- Can I get individual genotype information from browser.1000genomes.org?
How to download ENA files using aspera?
Answer:
The International Genome Sample Resource (IGSR) has stopped mirroring sequence files from the ENA but instead using the sequence.index files to point to the FTP location for the fastq file.
e.g ftp://ftp.sra.ebi.ac.uk/vol1/fastq/ERR008/ERR008901/ERR008901_1.fastq.gz
These files can also be downloaded using aspera. You will need to get the ascp program as described in how to download files using aspera
Then you will need to change the ENA FTP host to the ENA Aspera host.
This means you need to change the FTP url to something suitable for the ascp command:
e.g ftp://ftp.sra.ebi.ac.uk/vol1/fastq/ERR008/ERR008901/ERR008901_1.fastq.gz
becomes
fasp@fasp.sra.ebi.ac.uk:/vol1/fastq/ERR008/ERR008901/ERR008901_1.fastq.gz
You aspera command would need to look like
ascp -i bin/aspera/etc/asperaweb_id_dsa.openssh -Tr -Q -l 100M -L- fasp@fasp.sra.ebi.ac.uk:/vol1/fastq/ERR008/ERR008901/ERR008901_1.fastq.gz ./
Further details
For further information, please contact info@1000genomes.org. Full documentation about how to use aspera to download files from the ENA please see their document Downloading sequence files
Related questions:
How to download files using aspera?
Answer:
Download Aspera
Aspera provides a fast method of downloading data. To use the Aspera service you need to download the Aspera connect software. This provides a bulk download client called ascp.
Command line
For the command line tool ascp, for versions 3.3.3 and newer, you need to use a command line like:
ascp -i bin/aspera/etc/asperaweb_id_dsa.openssh -Tr -Q -l 100M -P33001 -L- fasp-g1k@fasp.1000genomes.ebi.ac.uk:vol1/ftp/release/20100804/ALL.2of4intersection.20100804.genotypes.vcf.gz ./
For versions 3.3.2 and older, you need to use a command line like:
ascp -i bin/aspera/etc/asperaweb_id_dsa.putty -Tr -Q -l 100M -P33001 -L- fasp-g1k@fasp.1000genomes.ebi.ac.uk:vol1/ftp/release/20100804/ALL.2of4intersection.20100804.genotypes.vcf.gz ./
Note, the only change between these commands is that for newer versions of ascp asperaweb_id_dsa.openssh replaces asperaweb_id_dsa.putty. This change is noted by Aspera here. You can check the version of ascp you have using:
ascp --version
The argument to -i may also be different depending on the location of the default key file. The command should not ask you for a password. All the IGSR data is accessible without a password but you do need to give ascp the ssh key to complete the command.
Files on the ENA FTP
Some of the data we provide URLs for is hosted on the ENA FTP site. ENA provide information on using Aspera with their FTP site.
As an example of downloading a file from ENA, you could use a command line like:
ascp -i bin/aspera/etc/asperaweb_id_dsa.openssh -Tr -Q -l 100M -P33001 -L-
era-fasp@fasp.sra.ebi.ac.uk:/vol1/fastq/ERR008/ERR008901/ERR008901_1.fastq.gz ./
Key files
If you are unsure of the location of asperaweb_id_dsa.openssh or asperaweb_id_dsa.putty, Aspera provide some documentation on where these will be found on different systems.
Ports
For the above commands to work with your network’s firewall you need to open ports 22/tcp (outgoing) and 33001/udp (both incoming and outgoing) to the following EBI IPs:
- 193.62.192.6
- 193.62.193.6
- 193.62.193.135
If the firewall has UDP flood protection, it must be turned off for port 33001.
Browser
Our aspera browser interace no longer works. If you wish to download files using a web interface we recommend using the Globus interface we present. If you are previously relied on the aspera web interface and wish to discuss the matter please email us at info@1000genomes.org to discuss your options.
Further details
For further information, please contact info@1000genomes.org.
Related questions:
How many individuals were sequenced?
Answer:
The 1000 Genomes Project aims to sequenced 2504 individuals in total both low coverage whole genome sequencing and exome sequencing. Further samples added into the IGSR will increase this number.
Related questions:
How much sequence data has been generated for single individuals?
Answer:
NA12878 the CEU child from our high coverage trio represents our largest amount of sequence data with 4.2 Tbases of sequence, the majority of this sequence data is from 2008 and short read length (~36bp) so is not the highest quality we have. You can see how many bases we have sequenced for all our samples by looking in our sequence index file. The 25th column of this file is the base count in each fastq file.
Related questions:
Is there any functional annotation for the 1000 Genomes phase 1 data?
Answer:
As part of our phase 1 analysis we performed functional annotation of our phase 1 variants with respect to both coding and non-coding annotation from GENCODE and the ENCODE project respectively.
This functional annotation can be found in our phase 1 analysis results directory. We present both the annotation we compared the variants to and VCF files which contain the functional consequences for each variant.
Related questions:
There is a corrupt file on your ftp site.
Answer:
As many of our files are very large >5GB they can become corrupt during download.
Before emailing us to let us know about a problem file the first thing you need to check is the md5 checksum of your file to see if it matches our records in the current.tree file.
If the size and or the md5 checksum don’t match then you need to attempt to download the file again.
If the size of the file and the md5 check sum matches what is in the current.tree then please email info@1000genomes.org to let us know which file has a problem.
Related questions:
What Affy 6.0 genotype data do you have?
Answer:
Coriel has carried out Affy 6.0 genotyping on many of the samples which are part of the NHGRI Cell line catalog.
These files are available on our FTP site in release/20130502/supporting/hd_genotype_chip/
Related questions:
What are CRAM files?
Answer:
CRAM files are alignment files like BAM files. They represent a compressed version of the alignment. This compression is driven by the reference the sequence data is aligned to.
The file format was designed by the EBI to reduce the disk footprint of alignment data in these days of ever-increasing data volumes.
The CRAM files the 1000 genomes project distributes are lossy cram files which reduce the base quality scores using the Illumina 8-bin compression scheme as described in the lossy compression section on the cram usage page
There is a cram developers mailing list where the format is discussed and help can be found.
CRAM files can be read using many Picard tools and work is being done to ensure samtools can also read the file format natively.
Related questions:
What are the kgp identifiers?
Answer:
kgp identifiers were not created by the 1000 Genomes Project. We also do not maintain them. They were created by Illumina for their genotyping platform before some variants identified during the pilot phase of the project had been assigned rs numbers.
We do not possess a mapping of these identifiers to current rs numbers. As far as we are aware no such list exists.
Related questions:
What are the targets for your exon targetted pilot study?
Answer:
The exon targetted run is part of the pilot study which targetted 1000 genes in nearly 700 individuals using a custom array. The targets for this pilot can be found in the pilot_data/technical/reference directory.
Related questions:
What are the targets for your whole exome sequencing?
Answer:
The exome sequencing the 1000 Genomes project has undertaken is targetting the entirety of the CCDS gene set.
The targets used for the phase 1 data release of 1092 samples can be found in technical/reference/exome_pull_down_targets_phases1_and_2; the targets for phase 3 analysis can be found in technical/reference/exome_pull_down_targets/.
The phase 1 and 2 targets are intersections of the different technologies used and the CCDS gene list. For phase 3 we are using a union of two different pull-down lists: NimbleGen EZ_exome v1 and Agilent sure select v2
In phase 3 very little exome specific calling took place. Instead analysis groups called variants tending to use the low coverage and exome data together in an integrated manner.
Related questions:
What are your filename conventions?
Answer:
Our filename conventions depend on the data format being named. This issue is described in more detail in the three questions below.
Related questions:
What Axiom genotype data do you have?
Answer:
The Affymetrix Axiom Exome chip was used to genotype some samples for the phase 1 analysis. There are genotypes for 1248 1000 Genomes samples from the Affy 6 chip available in phase1/analysis_results/supporting/axiom_genotypes from the Axiom Exome ChIP
Related questions:
What capture technology did the Exome sequencing use?
Answer:
Different centres have used different pull-down technologies for the Exome sequencing done for the 1000 Genomes project.
Baylor College of Medicine used NimbleGen SeqCap_EZ_Exome_v2 for its Solid based exome sequencing. For its more recent Illumina based exome sequencing it used a custom array HSGC VCRome
The Broad Institute has used Agilent SureSelect_All_Exon_V2 (https://earray.chem.agilent.com/earray/ using ELID: S0293689).
The BGI used NimbleGen SeqCap EZ exome V1 for the phase 1 samples and NimbleGen SeqCap_EZ_Exome_v2 for phase 2 and 3 (the v1 files were obtained from BGI directly; they are discontinued from Nimblegen).
The Washington University Genome Center used Agilent SureSelect_All_Exon_V2 (https://earray.chem.agilent.com/earray/ using ELID: S0293689) for phase 1 and phase 2, and NimbleGen SeqCap_EZ_Exome v3 for phase 3
Related questions:
What is the Data Slicer?
Answer:
The Data Slicer is a web based tool in our browser which allows you to get subsections of our indexed VCF and BAM files.
Related questions:
What is the depth of coverage of your Phase1 variants?
Answer:
The Phase 1 integrated variant set does not report the depth of coverage for each individual at each site. We instead report genotype likelihoods and dosage. If you would like to see depth of coverage numbers you will need to calculate them directly.
The bedtools suite provides a method to do this.
genomeCoverageBed is a tool which can provide a bed file which specifies coverage for every base in the genome and intersectBed which will provide an intersection between two vcf/bed/bam files
These commands also require samtools, tabix and vcftools to be installed
An example set of commands would be
samtools view -b ftp://ftp.1000genomes.ebi.ac.uk/vol1/ftp/data/HG01375/alignment/HG01375.mapped.ILLUMINA.bwa.CLM.low_coverage.20120522.bam 2:1,000,000-2,000,000 | genomeCoverageBed -ibam stdin -bg > coverage.bg
This command gives you a bedgraph file of the coverage of the HG01375 bam between 2:1,000,000-2,000,000
tabix -h http://ftp.1000genomes.ebi.ac.uk/vol1/ftp/phase1/analysis_results/integrated_call_sets/ALL.chr2.integrated_phase1_v3.20101123.snps_indels_svs.genotypes.vcf.gz 2:1,000,000-2,000,000 | vcf-subset -c HG01375 | bgzip -c > HG01375.vcf.gz
This command gives you the vcf file for 2:1,000,000-2,000,000 with just the genotypes for HG01375
To get the coverage for all those sites you would use
intersectBed -a HG01375.vcf.gz -b coverage.bg -wb > depth_numbers.vcf
You can find more information about bed file formats please see the Ensembl File Formats Help
For more information you may wish to look at our documentation about data slicing
Related questions:
What is the difference between the analysis groups exome and exon targetted in the sequence index?
Answer:
The 1000 Genomes Project has run two different pull-down experiments. These are labelled as “exon targetted” and “exome”.
An exon targetted run is part of the pilot study which targetted 1000 genes in nearly 700 individuals. The targets for this pilot can be found in the pilot_data/technical/reference directory.
An exome run is part of the whole exome sequencing project which targetted the entirety of the CCDS gene set. The targets used for the phase 1 data release of 1092 samples can be found in technical/reference/exome_pull_down_targets_phases1_and_2; the targets for phase3 analysis can be found in technical/reference/exome_pull_down_targets/
Related questions:
What is the difference between the sequence.index and the analysis.sequence.index?
Answer:
The sequence.index file contains a list of all the sequence data produced by the project, pointers to the file locations on the ftp site and also all the meta data associated with each sequencing run.
For the phase 3 analysis the consortium has decided to only use Illumina platform sequence data with reads of 70 base pairs or longer. The analysis.sequence.index file contains only the active runs which match this criterion. There are withdrawn runs in this index. These runs are withdrawn because either: * They have insufficient raw sequence to meet our 3x non duplicated aligned coverage criteria for low coverage alignments. * After the alignment has been run they have failed our post alignment quality controls for short indels. * Contamination. * They do not meet our coverage criteria.
Since the alignment release based on 20120522, we have only released alignments based on the analysis.sequence.index
Related questions:
What is the difference between your data directory and the pilot_data/data directory?
Answer:
The data directory represents the most current up-to-date view of sequence and alignment data available for the project. We also have a frozen data set which represents the data which was aligned for the pilot project as published in Nature in 2010.
An important difference to note is that while the main project data is all mapped to the GRCh37 assembly the pilot project was mapped to the NCBI36 assembly so positions of variants and alignments reported in the pilot_data directory will be different to what you see in the main project and many genome browsers. Genome browser and variant database also display the 1000 Genomes variants re-mapped to GRCh38, so these will give different coordinates again; you can access GRCh37 on Ensembl and UCSC genome browsers.
Related questions:
What do the names of your fastq files mean?
Answer:
Our sequence files are distributed in gzipped fastq format
Our files are named with the SRA run accession E?SRR000000.filt.fastq.gz. All the reads in the file also hold this name. The files with _1 and _2 in their names are associated with paired end sequencing runs. If there is also a file with no number it is name this represents the fragments where the other end failed qc. The .filt in the name represents the data in the file has been filtered after retrieval from the archive. This filtering process is described in a README.
Related questions:
What do the names of your variant files mean and what format are the files?
Answer:
Our variant files are distributed in vcf format, a format initially designed for the 1000 Genomes Project which has seen wider community adoption.
The majority of our vcf files are named in the form:
| **<span style”color:red”>ALL</span>.<span style”color:blue”>chrN</span> | <span style”color:green”>wgs | wex</span>.<span style”color:orange”>2of4intersection</span>.<span style”color:violet”>20100804</span>.<span style”color:darkblue”>snps | indels | sv</span>.genotypes.<span style”color:darkred”>analysis_group</span>.vcf.gz**. |
This name starts with the <span style”color:red”>population</span> that the variants were discovered in, if ALL is specifed it means all the individuals available at that date were used. Then the <span style”color:blue”>region</span> covered by the call set, this can be a chromosome, <span style”color:green”>wgs</span> (which means the file contains at least all the autosomes) or <span style”color:green”>wex</span> (this represents the whole exome) and a <span style”color:orange”>description</span> of how the call set was produced or who produced it, the <span style”color:violet”>date</span> matches the sequence and alignment freezes used to generate the variant call set. Next a field which describes what <span style”color:darkblue”>type of variant</span> the file contains, then the <span style”color:darkred”>analysis group</span> used to generate the variant calls, this should be low coverage, exome or integrated and finally we have either sites or genotypes. A sites file just contains the first 8 columns of the vcf format and the genotypes files contain individual genotype data as well.
Release directories should also contain panel files which also describe what individuals the variants have genotypes for and what populations those individuals are from
Related questions:
What do your population codes like CEU or TSI mean?
Answer:
These codes represent our populations, each three letter code represents a different population, CEU means Northern Europeans from Utah and TSI means Tuscans from Italy. There is a summary of all these codes both in a readme on the ftp site and in the alternative question Which populations are part of your study?
Related questions:
What does the LDAF value mean in your phase1 VCF files?
Answer:
LDAF is an allele frequency value in the info column of our phase 1 VCF files.
Our standard AF values are allele frequencies rounded to 2 decimal places calculated using allele count (AC) and allele number (AN) values. LDAF is the allele frequency as inferred from the haplotype estimation.
You will note that LDAF does sometimes differ from the AF calculated on the basis of allele count and allele number. This generally means there are many uncertain genotypes for this site. This is particularly true close to the ends of the chromosomes.
Related questions:
What format are your alignments in and what do the names mean?
Answer:
All our alignment files are in BAM format, a standard alignment format which was defined by the consortium and has since seen wide community adoption. We also provide our alignments in CRAM Format
The bam file names look like:
<span style”color:red”>NA00000</span>.<span style”color:blue”>location</span>.<span style”color:green”>platform</span>.<span style”color:orange”>population</span>.<span style”color:violet”>analysis_group</span>.<span style”color:darkred”>YYYYMMDD</span>.bam
The bai index and bas statistics files are also named in the same way.
The name includes the <span style”color:red”>individual sample ID</span>, <span style”color:blue”>where the sequence is mapped to</span>, if the file has only contains mapping to a particular chromosome that is what the name contains otherwise, mapped means the whole genome mapping and unmapped means the reads which failed to map to the reference (pairs where one mate mapped and the other didn’t stay in the mapped file), <span style”color:green”>the sequencing platform</span>, <span style”color:orange”>the ethnicity of the sample</span> using our three letter population code, <span style”color:violet”>the sequencing strategy</span>. The <span style”color:darkred”>date</span> matches the date of the sequence used to build the bams and can also be found in the sequence.index filename.
Related questions:
What format are your sequence files?
Answer:
Our sequence files are distributed in FASTQ format
We use Sanger style phred scaled quality encoding
The files are all gzipped compressed and the format looks like this, with a 4 line repeating pattern
@ERR059938.60 HS9_6783:8:2304:19291:186369#7/2
GTCTCCGGGGGCTGGGGGAACCAGGGGTTCCCACCAACCACCCTCACTCAGCCTTTTCCCTCCAGGCATCTCTGGGAAAGGACATGGGGCTGGTGCGGGG
+
7?CIGJB:D:-F7LA:GI9FDHBIJ7,GHGJBKHNI7IN,EML8IFIA7HN7J6,L6686LCJE?JKA6G7AK6GK5C6@6IK+++?5+=<;227*6054
Related questions:
What High Density Genotyping information do you have?
Answer:
The 1000 genomes project has multiple sets of high density genotype information on both Illumina and Affymetrix Platforms. Please see the below answers for more info.
Related questions:
What is the HLA Diversity in 1000 Genomes Samples?
Answer:
HLA diversity is not something which was studied by the 1000 Genomes Project directly, however, groups have looked at the HLA diversity of the samples in the 1000 Genomes Project.
2018 data
The most recent of these studies was published by Laurent Abi-Rached, Julien Paganini and colleagues in 2018 and covers 2,693 samples from the work of the 1000 Genomes Project. Details of the study and data used in this work are available via the publication and the HLA types are available on our FTP site at ftp://ftp.1000genomes.ebi.ac.uk/vol1/ftp/data_collections/HLA_types/.
2014 data
The FTP site also hosts data from an earlier study by Pierre-Antoine Gourraud, Jorge Oksenberg and colleages at UCSF who carried out an HLA typing assay on DNA sourced from Coriell for 1000 Genomes samples. This earlier study looks at only the 1,267 samples that were available at that time.
The earlier work assessing HLA Diversity is publised in “HLA diversity in the 1000 Genome Dataset”, with data available from the 1000 Genomes FTP site in ftp://ftp.1000genomes.ebi.ac.uk/vol1/ftp/technical/working/20140725_hla_genotypes/.
Related questions:
What library insert sizes where used in the 1000 genomes project?
Answer:
The project has generally used short insert libraries between 100 and 600bp for Illumina sequence data. For SOLiD and 454 sequence data you will see a wider variety of insert sizes. The insert sizes are reported in both the sequence.index file and the bas files. The sequence index file contains the insert size reported to the SRA when the data was submitted, the bas files contain the mean insert size based on the alignment and the standard deviation from that mean.
Related questions:
What Omni genotype data do you have?
Answer:
Both the Sanger Institute and the Broad Institute have carried on genotyping of 1000 Genomes and HapMap samples on the Omni Platform. The most recent set of Omni genotypes can be found in the phase 3 release directory release/20130502/supporting/hd_genotype_chip/ These contain GRCh37 based vcf files for the chip, and normalised and raw intensity files.
The ShapeIt2 scaffolds for these data, which were used in the phase 3 haplotype refinement project, can also be found in the phase 3 supporting directory release/20130502/supporting/shapeit2_scaffolds/
Related questions:
What is a panel file?
Answer:
All our variant call releases since 20100804 have come with a panel file. This file lists all the individuals who are part of the release and the population they come from.
This is a tab delimited file which must have sample and population in its first two columns; some files may then have subsequent columns which describe additional information like which super population a sample comes from or what sequencing platforms have been used to generate sequence data for that sample.
The panel files have names like integrated_call_samples.20101123.ALL.panel or integrated_call_samples_v2.20130502.ALL.panel
These panel files can be used by our browser tools, the Data Slicer, Variant Pattern Finder and vcf to ped converter to establish population groups for filtering
Related questions:
What read lengths were used by the project?
Answer:
As the project started sequencing in 2008 it holds a wide range of read lengths, the Illumina and SOLiD data range between 25bp to 160bp read lengths. Our sequence index file report read and base counts for each fastq file which can be used to find this out more precisely. For the final analysis phase of the project only Illumina data which is 70bp or longer was used and where required samples were sequenced again to match this criterion.
Related questions:
What is a sequence index file?
Answer:
We describe our sequence meta data in sequence index files. The index for data from the 1000 Genomes Project can be found in the 1000 Genomes data collection directory. Additional indices are present for data in other data collections. Our old index files which describe the data used in the main project can be found in the historical_data directory
Sequence index files are tab delimited files and frequently contain these columns:
| Column | Title | Description |
|---|---|---|
| 1 | FASTQ_FILE | path to fastq file on ftp site or ENA ftp site |
| 2 | MD5 | md5sum of file |
| 3 | RUN_ID | SRA/ERA run accession |
| 4 | STUDY_ID | SRA/ERA study accession |
| 5 | STUDY_NAME | Name of study |
| 6 | CENTER_NAME | Submission centre name |
| 7 | SUBMISSION_ID | SRA/ERA submission accession |
| 8 | SUBMISSION_DATE | Date sequence submitted, YYYY-MM-DD |
| 9 | SAMPLE_ID | SRA/ERA sample accession |
| 10 | SAMPLE_NAME | Sample name |
| 11 | POPULATION | Sample population, this is a 3 letter code as defined in README_populations.md |
| 12 | EXPERIMENT_ID | Experiment accession |
| 13 | INSTRUMENT_PLATFORM | Type of sequencing machine |
| 14 | INSTRUMENT_MODEL | Model of sequencing machine |
| 15 | LIBRARY_NAME | Library name |
| 16 | RUN_NAME | Name of machine run |
| 17 | RUN_BLOCK_NAME | Name of machine run sector (This is no longer recorded so this column is entirely null, it was left in so as not to disrupt existing sequence index parsers) |
| 18 | INSERT_SIZE | Submitter specified insert size |
| 19 | LIBRARY_LAYOUT | Library layout, this can be either PAIRED or SINGLE |
| 20 | PAIRED_FASTQ | Name of mate pair file if exists (Runs with failed mates will have a library layout of PAIRED but no paired fastq file) |
| 21 | WITHDRAWN | 0/1 to indicate if the file has been withdrawn, only present if a file has been withdrawn |
| 22 | WITHDRAWN_DATE | This is generally the date the file is generated on |
| 23 | COMMENT | comment about reason for withdrawal |
| 24 | READ_COUNT | read count for the file |
| 25 | BASE_COUNT | basepair count for the file |
| 26 | ANALYSIS_GROUP | the analysis group of the sequence, this reflects sequencing strategy. For 1000 Genomes Project data, this includes low coverage, high coverage, exon targeted and exome to reflect the two non low coverage pilot sequencing strategies and the two main project sequencing strategies used by the 1000 Genomes Project. |
Related questions:
What Sequencing Platforms were used for the 1000 Genomes Project?
Answer:
For the final phase of the 1000 Genomes Project only Illumina was used for the sequencing. The phase 1 effort also included ABI SOLiD sequence and the pilot project contained 454 sequence data.
Related questions:
What strand are the variants in your VCF file on?
Answer:
All the variants in both our VCF files and on the browser are always reported on the forward strand.
Related questions:
What structural variant data is available for the project?
Answer:
The project has two releases of structural variation. The pilot paper data directory contains vcf files for deletions, mobile element insertions, tandem duplications and novel sequence both for the low coverage and trio pilot studies. Our phase1 release integrated release contains deletions together with the SNPs and short indels.
Related questions:
What tools can I use to download 1000 Genomes data?
Answer:
The 1000 Genomes data is available via ftp, http and Aspera. Any standard tool like wget or ftp should be able to download from our ftp or http mounted sites. To use Aspera you need to download their client.
Related questions:
What version of VCF are your VCF files in?
Answer:
The VCF files on our site cover a wide variety of different versions but our most recent release VCF files are in format version 4.1
Related questions:
What was the source of the DNA for sequencing?
Answer:
The early samples were taken from the HapMap project and these all sourced their dna from cell line cultures. Some of the more recent libraries have been produced from blood.
Our sample spreadsheet has annotation about the EBV coverage and the annotated sample source of the sequencing data in columns 59 and 60 of the 20130606_sample_info.txt
This spreadsheet gives the aligned coverage of EBV and then the annotation if Coriell stated the sample was sources from blood. Please note some samples were sequenced both using sample from blood and LCL transformed cells but this data was not analysed independently so the EBV coverage will be high. Also some samples with very low EBV coverage (ie ~1x) may be from blood but just indicate an endogenous infection of EBV in the individual sampled.
Related questions:
Where are the alignment files for the exon targetted individuals?
Answer:
The alignment files which were used as part of the pilot project are found under ftp://ftp.1000genomes.ebi.ac.uk/vol1/ftp/pilot_data/data. These are all aligned to NCBI36.
There are also GRCh37 alignments available for both the high coverage individuals (ftp://ftp.1000genomes.ebi.ac.uk/vol1/ftp/technical/pilot2_high_cov_GRCh37_bams/) and the exon targetted individuals (ftp://ftp.1000genomes.ebi.ac.uk/vol1/ftp/technical/pilot3_exon_targetted_GRCh37_bams/)
Please be aware much of the sequence data these alignments are based on is very short read data (36bp) and was generated a long time ago (~2008) so may not reflect current sequencing data.
A more modern data set for the CEU trio is available from Illumina on their Platinum Genomes page.
Where are the pilot structural variants archived?
Answer:
The longer structural variants predicted as part of the pilot project were submitted to the DGVa and given the accession estd59.
Related questions:
Where are the SNPs for the X/Y/Mitochondrial chr?
Answer:
Chromosome X, Y and MT variants are available for the phase 3 variant set. The chrX calls were made in the same manner as the autosome variant calls and as such are part of the integrated call set and include SNPs, indels and large deletions, note the males in this call set are show as haploid for any chrX SNP not in the Pseudo Autosomal Region (PAR). The chrY and MT calls were made independently. Both call sets are presented in an integrated file in the phase3 FTP directory, chrY and chrMT. ChrY has snps, indels and large deletions. ChrMT only has snps and indels. For more details about how these call sets were made please see the phase 3 paper.
Related questions:
Where are your alignment files located?
Answer:
Our main alignment files are located in our data directory. Our mapped bams contain reads which aligned to the whole genome.
You can find an index of our alignments in our alignment.index file. There are dated versions of these files and statistics surrounding each alignment release in the alignment_indicies directory. Please note with few exceptions we only keep the most recent QC passed alignment for each sample on the ftp site.
We also have frozen versions of the alignments use for both the pilot and the phase 1 analyses in different directories on the ftp site. Please note the that the pilot alignments are mapped to NCBI36 rather than GRCh37 like all other alignments on the ftp site.
Related questions:
Where are your reference data sets?
Answer:
Our reference data sets can be found in technical/reference/ and this includes items like the reference genome, ancestral alignments and standard annotation sets.
There is also a frozen version of the reference data used for the pilot project available in pilot_data/technical/reference
Related questions:
Where are your sequence files located?
Answer:
Our sequence files are distributed in fastq format and can be found under the data directory of the ftp site, here there is a directory per individual which then contains all the sequence data we have for that individual aswell as all the alignment data we have.
We also distribute meta data for all our sequencing runs in a sequence.index file which is described in a README on the ftp site.
Related questions:
Where are your variant files located?
Answer:
Our variant files are released via our release directory in directories named for the sequence index freeze they are based on.
You may find information about the final 1000 Genomes release in contained in 20130502 and is described in where is your most recent release?
A stable earlier release based on 1092 unrelated samples is phase 1 data release, it can be found under phase1/analysis_results/integrated_call_sets. The phase 1 data set contains information on all autosomes and chrX, Y and chrMT. The phase 1 publication is based on this data set.
The pilot release represents results obtained in the three pilot studies of the project (low coverage, high coverage trios and exome). The release data can be found at here. The publication about the findings of pilot studies is in this pdf.
You may also find variant files in our technical/working directory but please be aware these are experimental files which represent a work in progress and should always be treated with caution
Related questions:
Where can I get consequence annotations for the 1000 genome variants?
Answer:
The final 1000 Genomes phase 3 analysis calculated consequences based on GENCODE annotation and this can be found in the directory: release/20130502/supporting/functional_annotation/
Ensembl can also provides consequence information for the variants. The variants that are loaded into the Ensembl database and have consequence types assigned and displayed on the Variation view. Ensembl can also offer consequence predictions using their Variant Effect Predictor (VEP).
Please note the phase 3 annotations and the Ensembl annotations visible via the browser due to using different versions of gene and non coding annotation.
Related questions:
Where is the most recent release?
Answer:
Our main releases are contained in the release directory on our ftp site.
These directories are dated for the sequence index freeze the analysis products are based on.
Our most current recent release is the phase 3 release in 20130502. This represents an integrated set of SNPs, indels, MNPs, long insertions and deletions, copy number variations, and other types of structural variations discovered and genotyped in 2504 unrelated individuals.
Related questions:
Which reference assembly do you use?
Answer:
The reference assembly the 1000 Genomes Project has mapped sequence data to has changed over the course of the project.
For the pilot phase we mapped data to NCBI36. A copy of our reference fasta file can be found on the ftp site.
For the phase 1 and phase 3 analysis we mapped to GRCh37. Our fasta file which can be found on our ftp site called human_g1k_v37.fasta.gz, it contains the autosomes, X, Y and MT but no haplotype sequence or EBV.
Our most recent alignment release was mapped to GRCh38, this also contained decoy sequence, alternative haplotypes and EBV. It was mapped using an alt aware version of BWA-mem. The fasta files can be found on our ftp site
Related questions:
Which samples are you sequencing?
Answer:
There is a list of samples who are part of the project available from this spreadsheet. There is also a pedigree file available from ftp://ftp.1000genomes.ebi.ac.uk/vol1/ftp/technical/working/20130606_sample_info/20130606_g1k.ped
Please note this spreadsheet does list samples who are related to the ones we are sequencing but aren’t themselves being sequenced. If a sample has no data in the Total LC or Total E Sequence columns it means it was not sequenced for the main project
Related questions:
Why are the coordinates of your pilot variants different to what is displayed in Ensembl or UCSC?
Answer:
The pilot data for the 1000 genomes project was all mapped to NCBI36/hg18 build of the human assembly. When the data was been loaded into dbSNP it was mapped to GRCh37/hg19 which is accessible from both Ensembl and UCSC but this does mean that the coordinates from the pilot data on the 1000 Genomes ftp site will be different to the coordinates presented in Ensembl and UCSC.
You can also view 1000 Genomes variants mapped to GRCh38 on Ensembl and UCSC.
Related questions:
Why are there chr 11 and chr 20 alignment files, and not for other chromosomes?
Answer:
The chr 11 and chr 20 alignment files are put in place to give the 1000 Genomes analysis group a small section of the genome to run test analyses on before committing to a particular strategy to run across the whole genome. Everything in the chr 11 and chr 20 files is also represented in the mapped bam file. To get a complete view of what data we aligned you only need to download the mapped and unmapped bams, the chr 11 and chr 20 bams are there as a convenience to the analysis group.
Related questions:
Why are there more than one set of fastq files associated with an individual?
Answer:
Many of our individuals have multiple fastq files. This is because many of our individual were sequenced using more than one run of a sequencing machine.
Each set of files named like ERR001268_1.filt.fastq.gz, ERR001268_2.filt.fastq.gz and ERR001268.filt.fastq.gz represent all the sequence from a sequencing run.
When a individual has many files with different run accessions (e.g ERR001268), this means it was sequenced multiple times. This can either be for the same experiment, some centres used multiplexing to have better control over their coverage levels for the low coverage sequencing, or because it was sequenced using different protocols or on different platforms.
For a full description of the sequencing conducted for the project please look at our sequence.index file
Related questions:
Why do some of your vcf genotype files have genotypes of ./. in them?
Answer:
Our August 2010 call set represents a merge of various different independent call sets. Not all the call sets in the merge had genotypes associated with them, as this merge was carried out using a predefined rules which has led to individuals or whole variant sites having no genotype and this is described as ./. in vcf 4.0. In our November 2010 call and all subsequent call sets all sites have genotypes for all individuals for chr1-22 and X.
Related questions:
Why does a tabix fetch fail?
Answer:
There are two main reasons a tabix fetch might fail.
All our VCF files using straight intergers and X/Y for their chromosome names in the Ensembl style rather than using chr1 in the UCSC style. If you request a subsection of a VCF file using a chromosome name in the style chrN as shown below it will not work.
tabix -h ftp://ftp.1000genomes.ebi.ac.uk/vol1/ftp/release/20100804 ALL.2of4intersection.20100804.genotypes.vcf.gz chr2:39967768-39967768
Also tabix does not fail when streaming remote files but instead just stops streaming. This can lead to incomplete lines with final rows with unexpected numbers of columns when trying to stream large sections of the file. The only way to avoid this is to download the file and work with it locally.
Related questions:
Why isn't my SNP in browser.1000genomes.org?
Answer:
Ensembl and UCSC Genome Browser both import their variant data from dbSNP. When new 1000 Genomes variants have been released it can take some time for them to be accessioned by dbSNP and make their way to the browsers.
When this happens we try to ensure there is a version of our own browser which displays the data in the meantime. Both Ensembl and UCSC support attaching VCF files to them for visualisation
Related questions:
Why isn't a SNP in dbSNP or HapMap?
Answer:
The 1000 Genomes Project submits all its variants to archives like dbSNP or the DGVa. If it hasn’t yet made it to dbSNP this means it is likely to be a new site which we haven’t yet submitted. There may also be some old sites which we subsequently discover to be false discoveries which we then suppress.
As far as our overlap with the HapMap site list goes, The majority of HapMap SNPs are found in the 1000 Genomes Project, there will be a small number of sites we fail to find using next generation sequencing but most sites from HapMap which aren’t found by the 1000 Genomes Project will be false discoveries by HapMap. There are a lot of SNPs from the 1000 Genomes Project and other next generation sequencing projects which won’t be part of HapMap as HapMap is based on an older genotyping technology when such rapid variant discovery using sequencing was not possible.
Related questions:
Why is the sequence data distributed in 2 or 3 files labelled SRR_1, SRR_2 and SRR?
Answer:
We distribute our fastq files for our paired end sequencing in 2 files, mate1 is found in a file labelled _1 and mate2 is found in the file labelled _2. The files which do not have a number in their name are singled ended reads, this can be for two reasons, some sequencing early in the project was singled ended also, as we filter our fastq files as described in our README if one of a pair of reads gets rejected the other read gets placed in the single file.